对药物分子进行氟代修饰,可以改善药物分子的代谢稳定性和血脑屏障通透性。近年来,德国马克斯•普朗克煤炭研究所的Tobias Ritter教授(点击查看介绍)课题组致力于发展芳环的氟化反应,因此直接的碳氢键官能团化反应就成为一种极具优势的策略。噻蒽及其衍生物能够形成稳定的自由基,从而与芳环发生自由基加成反应。然而,常见的自由基对芳环的加成反应往往选择性不高,因此寻找电荷亲和度高(即对富电子位点选择性高)的自由基阳离子是较好的解决策略,其中噻蒽(或二苯并噻吩)的三氟甲磺酸锍盐可与富电子芳环发生反应。另外,根据芳环电性的差异性,所使用的噻蒽(或二苯并噻吩)也有所不同。
(1)芳环的官能团化
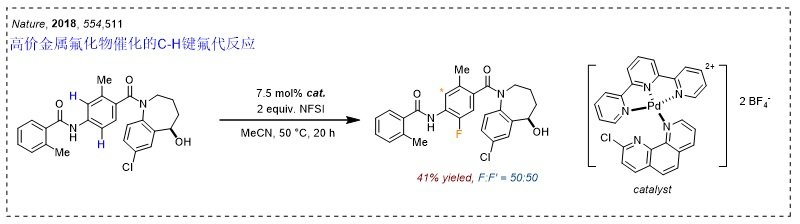
2018年,该课题组报道了高价金属氟化物中间体催化的芳环碳氢键氧化氟化反应(Nature, 2018, 554, 511,点击阅读详细)。然而,该策略对选择性的控制不能尽如人意。
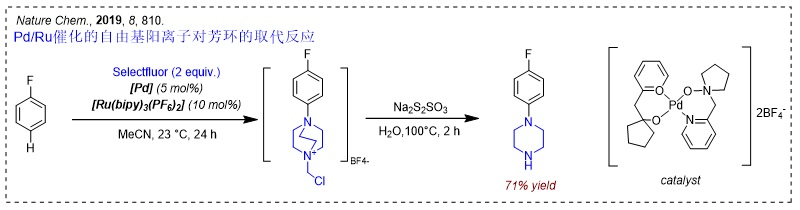
此前的2016年,他们发现高亲电自由基阳离子TEDA2+·能够实现芳环的区域选择性,并发展了Pd/Ru双金属催化的芳环的C-H键官能团化反应(Nat. Chem., 2016, 8, 810, 点击阅读详细)。然而,该反应只能用Selectfluor试剂将可预测位置转化成铵。尽管如此,通过自由基阳离子来实现芳环的选择性官能团化已初现端倪。
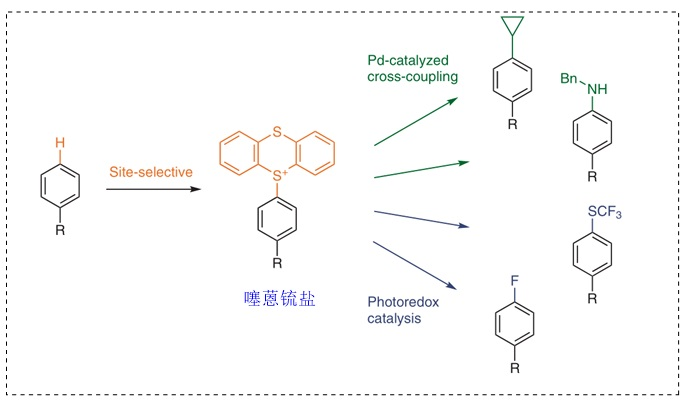
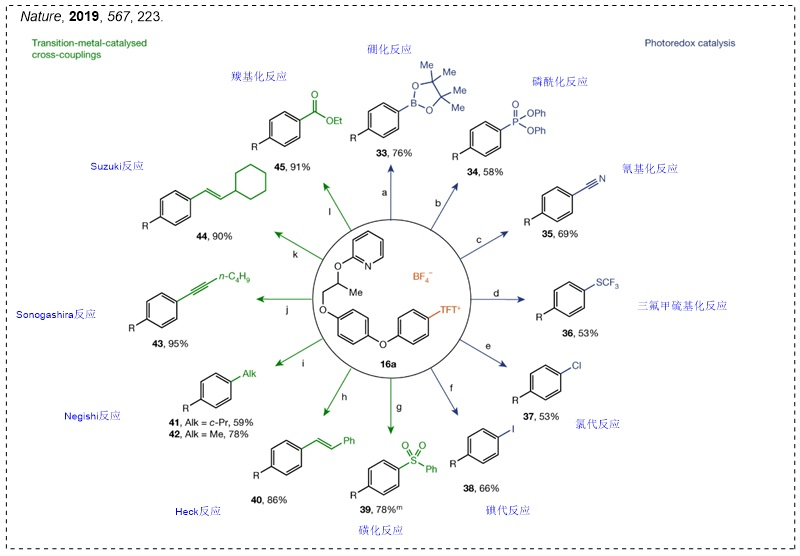
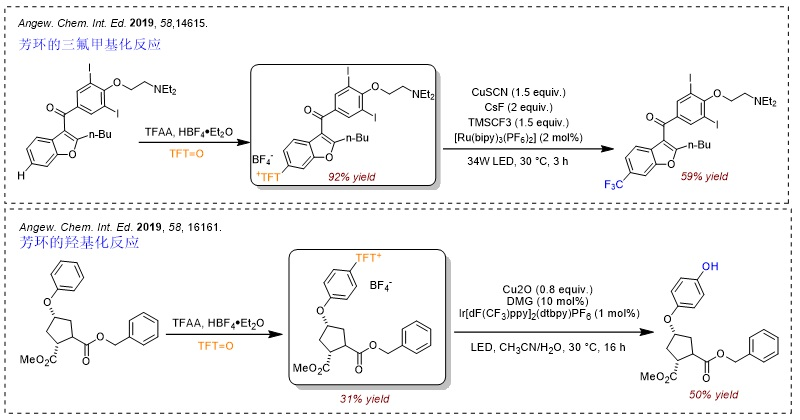
随后,他们基于噻蒽鎓盐产生的稳定噻蒽阳离子自由基,成功地实现了芳基C-H键的选择性官能团化反应(Nature, 2019, 567, 223, 点击阅读详细),并且得到的产物还能进行后续的衍生化,例如:羟基化、硼化、溴化、氯化、烯基化、氟化、烷基化等。下面对基于锍盐的反应做一个简单的介绍:
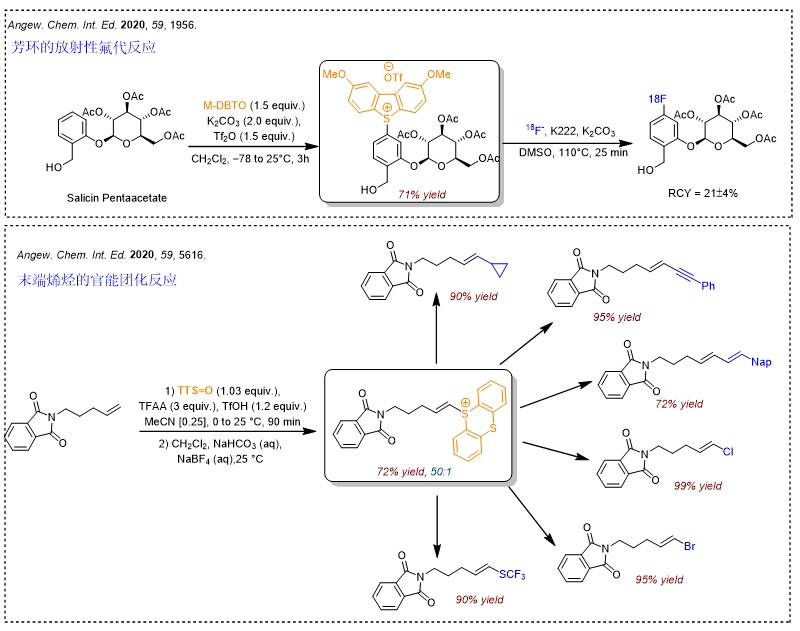
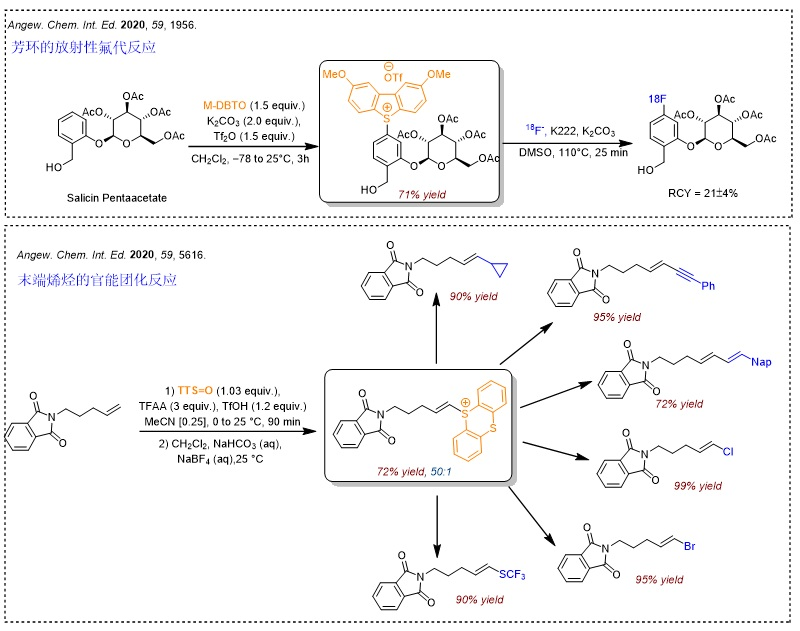
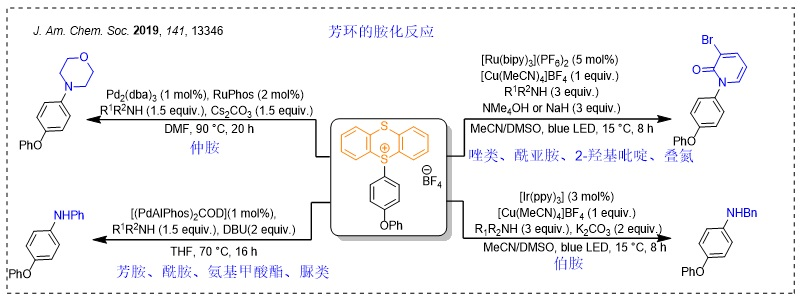
噻蒽锍盐的胺化反应是另一类较为重磅的反应。常用的氨基化反应(如Buchwald-Hartwig偶联反应)往往需要以卤化物作为前体,但新方法直接利用锍盐进行C-H键官能团化,更为直接和便捷。事实上,锍盐主要选择富电子的位点进行反应,因此它仍然需要和其它胺化反应互为补充。
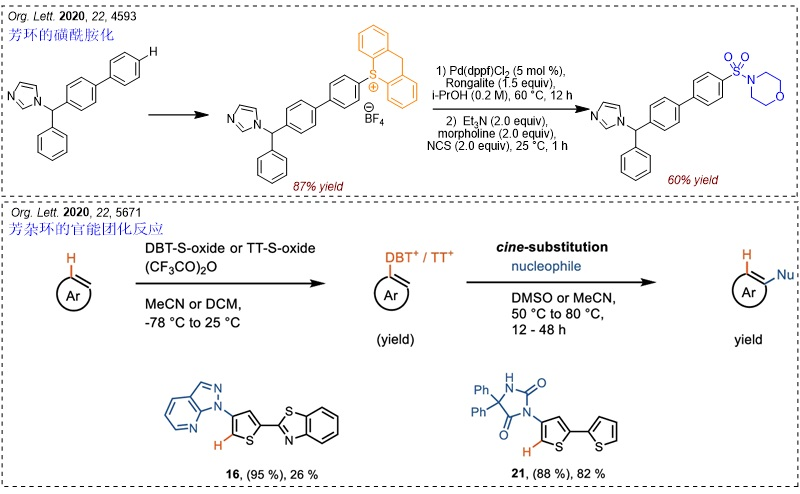
此外,鉴于锍盐自由基阳离子的稳定性以及形成衍生物的选择性、离去活性,更多的取代反应也被开发出来,包括磺酰胺化、噻吩环的官能团化反应等。
(2)杂环的合成
继噻蒽亚砜被广泛应用于芳环的自由基加成反应-官能团化之后,Tobias Ritter课题组继续研究了该试剂在烯烃的官能团化反应中的应用。与末端烯烃的官能团化不同(Angew. Chem. Int. Ed., 2020, 59, 5616),该反应并不以杂Diels-Alder加成反应的形式进行,而是经光介导的氮自由基环化过程进行。
含氮杂环是药物化学中至关重要的砌块之一,据统计,FDA批准的药物分子中一半以上都含有至少一个含氮杂环。目前,构建含氮杂环化合物的重要方法之一是Buchwald-Hartwig反应,可将不同片段通过C-N键偶联来链接。更多样性的方法则是通过分子内的缩合、环化、取代等经典的杂环合成方法为主。另外,分子间的烯烃双官能团化反应是近几十年以来发展出来的新型杂环构建策略,但是受限于烯烃本身的性质,模块化的合成方法往往十分有限。得益于N-H键较高的键能(107 kal/mol,NH3),N中心自由基(nitrogen-centered radical,NCR)在进行分子内氢原子转移(HAT)过程中是一类活泼的反应物种,因此可以作为关键的中间体来构建新的C-N键。另外,NCR也可以作为亲电自由基,与富电子烯烃、芳烃加成形成相应的烷基胺或芳胺。实际上,该策略被广泛应用于烯烃的加氢胺化、氨氧化、氨氟化、碳胺化、氨叠氮化反应等。尽管如此,但是NCR前体所含有的磺酰基等用于增强其亲电能力的保护基也限制了其应用。近日,Tobias Ritter教授课题组利用硫亚胺(由二苯并亚砜与胺制备而成)为双功能N-自由基前体,在光氧化还原催化下实现了硫亚胺与烯烃的分子间环化反应,一步法构建了多种未保护的含氮杂环,如吗啉、哌嗪、二氢噁唑、二氢咪唑衍生物等。相关成果发表在Nature Chemistry 上。
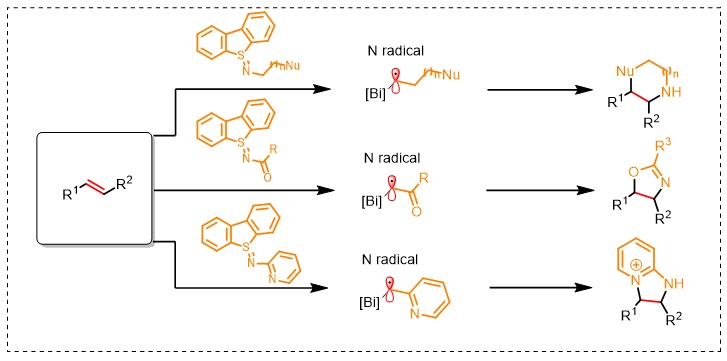
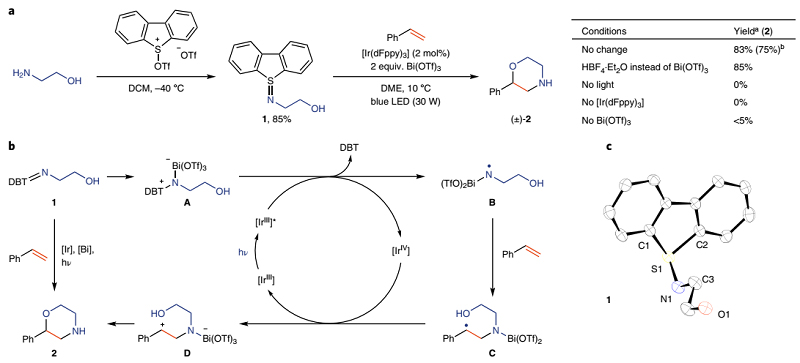
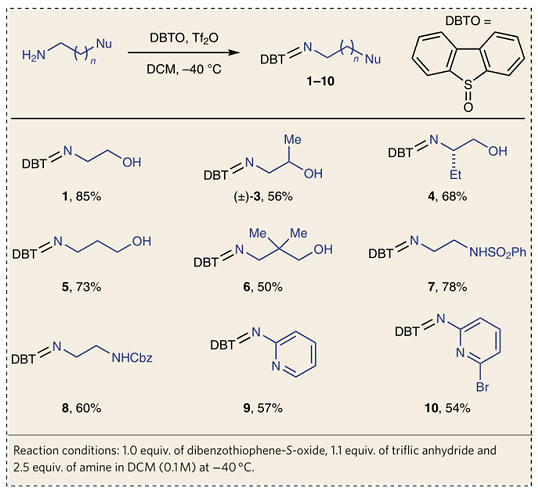
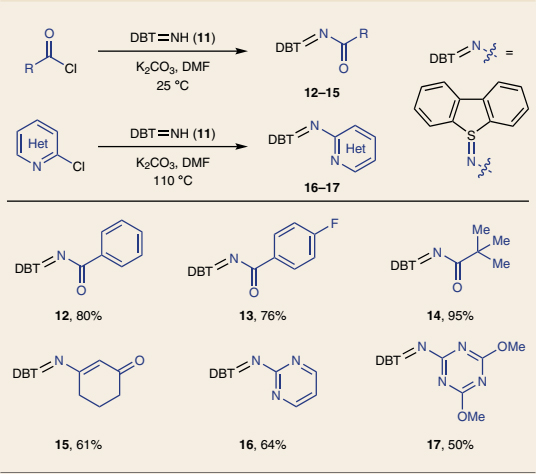
首先,作者从市售可得的氨基乙醇和三氟甲磺酸酐活化的二苯并噻吩-S-氧化物出发,一步法合成双功能N-自由基前体——硫亚胺1,再与苯乙烯在光氧化还原催化下进行反应便可得到相应的苯基吗啉2。尽管该反应需要化学计量的二苯并噻吩-S-氧化物,但是在环化后能重复使用。另外,对照实验表明光照和光氧化还原催化剂对该反应至关重要,并且Brønsted酸和Lewis酸均能加速环化反应,这可能是由于酸能与硫亚胺配位,从而有效地促进其与激发态光催化剂之间的单电子转移(SET)。需要指出的是,Brønsted酸往往会不可避免地诱发富电子苯乙烯的聚合反应,而Bi(OTf)3则能最大程度上促进该自由基环化过程。作者一步法合成了多种双功能硫亚胺试剂(3-10、12-17),大多都是固体并且能在室温下稳定存在至少3个月,产物6除外。
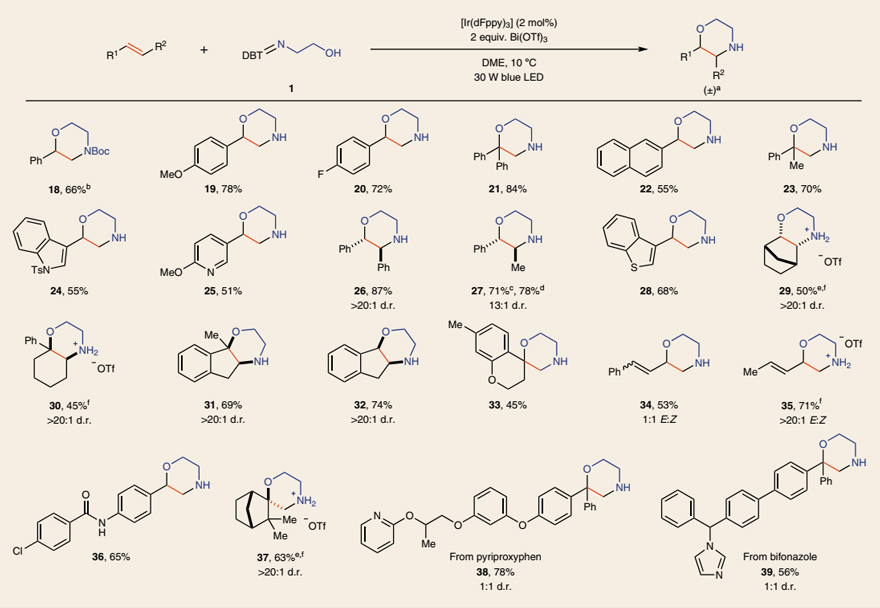
接下来,作者对该反应的底物范围进行了考察。苯乙烯及其衍生物(18-23、36)、杂芳基取代的烯烃(24、25、28)、1,2-二取代烯烃(26、27、29、32)、1,1-二取代烯烃(21、23、30、31、33、37)甚至环状烯烃(29-32)均能兼容该反应,以中等至较好的收率得到相应的吗啉产物,包括双环/三环吗啉、螺环吗啉。此外,对硫亚胺上的取代基稍加修改后,就能合成多种其它类型的杂环化合物,例如二取代吗啉(42、43)、七元氮氧杂环(44、45)、哌嗪(46、47)、二氢噁唑(50-52)、二氢咪唑衍生物(48、49、54、55)。有趣的是,当使用硫亚胺15进行反应时,仅以89%的收率得到四氢苯并呋喃53而非N-杂环,这可能是由于生成的烯胺 NCR 转化为更稳定的碳中心自由基,然后参与1,1-二苯乙烯的环化。最后,作者还以两步、50%的总收率合成了H1受体拮抗剂41,而先前的方法则需要5步并且收率仅为2%,这进一步凸显出该方法的合成价值。
然而作者也提到该方法对非活化的烯烃如3-苯丙烯、链状烯烃、丙烯酸甲酯则是无能为力的。因为在没有芳环稳定自由基的情况下,该自由基不能够稳定的参与氧化过程,从而不能进一步的发生成环反应。
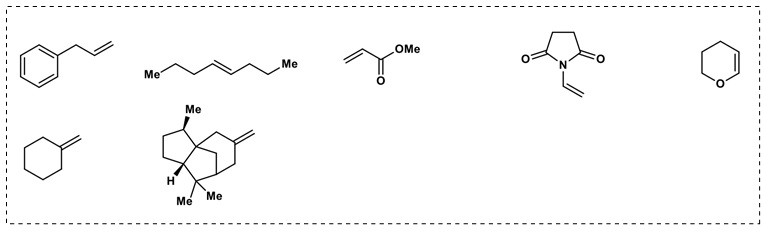
为了进一步探究反应机理,作者进行了一系列实验。硫亚胺1和Bi(OTf)3混合物(1:1)的循环伏安图显示出一个新的电位峰,作者将其归因于1-Bi(OTf)3加合物A,并通过紫外-可见光谱(326 nm处有吸收峰)证实了其存在。另外,作者还在最优条件下进行了自由基钟实验,分别得到开环产物56 和环化产物 57,进一步证实了NCR的存在。在此基础上,作者提出了可能的反应机理:首先,硫亚胺1和Bi(OTf)3形成1-Bi(OTf)3加合物A,A在光催化的作用下产生N中心自由基B。随后,B与烯烃进行加成得到相应的碳自由基C,再被[IrIV]氧化为相应的碳正离子D,最后经分子内亲核进攻得到相应的含氮杂环2。
总结
Tobias Ritter教授在光氧化还原催化下实现了硫亚胺与烯烃的分子间环化反应,该策略不仅能避免在自由基环化过程中所使用的保护基,而且该反应中的硫亚胺试剂会进一步丰富有机化学、药物合成化学的杂环合成策略。推而广之,Tobias Ritter组所发展的一系列基于二苯并噻吩、噻蒽类试剂,在C-H键官能团化上表现出卓越的性能,尤其是在药物分子的后期修饰上,这将成为药物化学家关注的新焦点。
Bifunctional sulfilimines enable synthesis of multiple N-heterocycles from alkenes
Qiang Cheng, Zibo Bai , Srija Tewari, Tobias Ritter
Nat. Chem., 2022, 14, 898-904, DOI: 10.1038/s41557-022-00997-y